This page gives a description of CTX, the symptoms, the diagnosis, and other details about the disease. This text is taken from the genetests.com website and is a very comprehensive article about the disease.
Here is a great PDF that summarizes the various symptoms and states of CTX:
Here is an article about Ashley(aka ‘Betty’) written by Dr. Charles Bock, Ashley’s Ophthalmologist and Dr. Arif Khan, an Ophthalmologist who practices in Saudi Arabia:
Ophthalmologists and CTX – Making the Diagnosis
Summary
Disease characteristics. Cerebrotendinous xanthomatosis (CTX) is a lipid storage disease characterized by infantile-onset diarrhea, childhood-onset cataract, adolescent-to-young-adult-onset tendon xanthomas, and adult-onset progressive neurologic dysfunction (dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, and seizures). Chronic diarrhea from infancy may be the earliest clinical manifestation. In about 75% of affected individuals, cataracts are the first finding, often appearing in the first decade of life. Xanthomas appear in the second or third decade. Xanthomas occur on the Achilles tendon, the extensor tendons of the elbow and hand, the patellar tendon, and the neck tendons. Xanthomas have been reported in the lung, bones, and central nervous system. Some individuals show mental impairment from early infancy, whereas the majority have normal or only slightly subnormal intellectual function until puberty. Dementia with slow deterioration in intellectual abilities in the 20s occurs in over 50% of individuals. Neuropsychiatric symptoms such as behavioral changes, hallucinations, agitation, aggression, depression, and suicide attempts may be prominent. Pyramidal signs (i.e., spasticity) and/or cerebellar signs are almost invariably present between 20-30 years of age. Other findings include extrapyramidal manifestations (dystonia and atypical parkinsonism), seizures, and peripheral neuropathy.
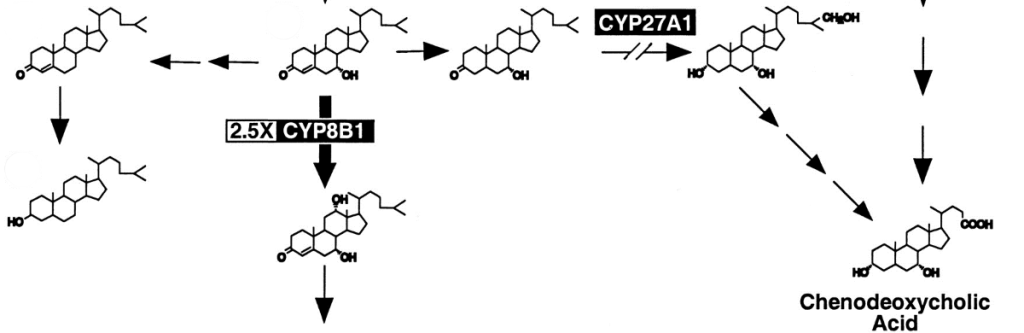
Diagnosis/testing. CTX is caused by deficiency of the mitochondrial enzyme sterol 27-hydroxylase with resulting cholestanol and cholesterol accumulation in virtually every tissue. The laboratory abnormalities that distinguish CTX from other conditions with xanthomas include: high plasma and tissue cholestanol concentration, normal-to-low plasma cholesterol concentration, and increased concentration of bile alcohols and their glyconjugates in bile, urine, and plasma. CYP27A1 is the only gene known to be associated with cerebrotendinous xanthomatosis. Molecular genetic testing of the CYP27A1 gene is available on a research basis only.
Genetic counseling . CTX is inherited in an autosomal recessive manner. At conception, the sibs of an affected individual have a 25% chance of being affected , a 50% chance of being asymptomatic carriers , and a 25% chance of being unaffected and not carriers .Prenatal testing is not currently available except possibly through laboratories offering custom prenatal testing .
Diagnosis
Clinical Diagnosis
Cerebrotendinous xanthomatosis (CTX), a lipid storage disease, is suspected in individuals with infantile-onset diarrhea, childhood-onset cataract, adolescent-to young-adult-onset tendon xanthomas, and adult-onset progressive neurologic dysfunction (dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, and seizures).
MRI. MRI shows bilateral hyperintensity of the dentate nuclei and cerebral and cerebellar white matter.
Testing
CTX is caused by deficiency of the mitochondrial enzyme sterol 27-hydroxylase with resulting cholestanol and cholesterol accumulation in virtually every tissue.
Biochemical testing. The main laboratory abnormalities that distinguish CTX from other conditions with xanthomas:
- High plasma and tissue cholestanol concentration
- Normal to low plasma cholesterol concentration
- Markedly decreased formation of chenodeoxycholic acid resulting from impaired primary bile acid synthesis
- Increased concentration of bile alcohols and their glyconjugates in bile, urine, and plasma
- Increased concentration of cholestanol and apolipoprotein B in cerebrospinal fluid (CSF) resulting from changes in the blood-brain barrier
Genetically Related Disorders
No other phenotypes are known to be associated with mutations in CYP27A1 .
Clinical Description
Cerebrotendinous xanthomatosis (CTX) is suspected in individuals with infantile-onset diarrhea, childhood-onset cataract, adolescent-to-young-adult-onset tendon xanthomas, and adult-onset progressive neurologic dysfunction (dementia, psychiatric disturbances, pyramidal and/or cerebellar signs, and seizures). Intrafamilial variability is considerable [ Dotti et al 1996 ; Nagai et al 1996 ; Verrips, Hoefsloot et al 2000 ].
A distinction can be made between systemic signs and neurological signs, described in detail below.
Systemic Signs
Enterohepatic system. Chronic diarrhea from infancy may be the earliest clinical manifestation of CTX [ Cruysberg et al 1991 , Cruysberg 2002 ]. Gallstones have been reported on occasion.
Eye. In about 75% of affected individuals, cataracts are the first finding, often appearing in the first decade of life. However in 25% of individuals, cataracts are first observed after the age of 40 years. Cataracts may be visually significant opacities requiring lensectomy or visually insignificant cortical opacities. The appearance can include irregular cortical opacities, anterior polar cataracts, and dense posterior subcapsular cataracts [ Cruysberg et al 1995 ]. Other findings include palpebral xanthelasmas [ van Bogaert et al 1937 , Philippart & van Bogaert 1969 ], optic nerve atrophy [ Schimschock et al 1968 ], and proptosis [ Morgan et al 1989 ]. In 13 individuals reported by Dotti et al ( 2001 ) ranging in age from 32-54 years, all had cataracts, about 50% had optic disk paleness, 30% had signs of premature retinal senescence with retinal vessel sclerosis, 15% had cholesterol-like deposits along vascular arcades, and 15% had myelinated nerve fibers.
Xanthomas. These appear in the second or third decade. In addition to the classic xanthomatas of the Achilles tendon, xanthomas also occur on the extensor tendons of the elbow and hand, the patellar tendon, and the neck tendons. Xanthomas have been reported in the lung, bones, and central nervous system (CNS).
Cardiovascular system. Premature atherosclerosis and coronary artery disease have been reported [ Schimschock et al 1968 , Fujiama et al 1991 , Kerleau et al 1993 ]; however, occurrence of atherosclerosis in CTX homozygotes has been attributed to factors other than mutation of CP27A1 [ Leitersdorf et al 1994 ]. Dotti et al ( 1998 ) described lipomatous hypertrophy of the atrial septum.
Skeleton. Bone involvement is characterized by granulomatous lesions in the lumbar vertebrae and femur, osteopenia and increased risk of bone fractures, and impaired adsorption of radiocalcium, which improves with chenodeoxycholic acid treatment [ Berginer et al 1993 , Federico et al 1993 ]. Osteopenia is evident by total body densitometry in untreated individuals. Individuals may have marked thoracic kyphosis.
Endocrine abnormalities. Hypothyroidism has occasionally been reported [ Philippart & van Bogaert 1969 , Bouwes Bavinck et al 1986 , Idouji et al 1991 ].
Premature aging. Early-onset cataract, osteopenia with bone fractures and loss of teeth, atherosclerosis, and neurological impairment with dementia and/or parkinsonism, associated with the characteristic facies, suggest a generalized premature aging process [ Dotti et al 1991 ].
Histologic changes. Histologic liver findings include electron-dense amorphous material surrounded by smooth endoplasmic reticulum [ Salen et al 1978 ] and abnormalities in mitochondria with paracrystalline inclusions and increased number of peroxisomes [Federico et al 1989 ]. Histologically they are characterized by birefringent crystalline material surrounded by numerous multinucleate giant cells with foamy cytoplasm.
Neurologic Signs
Mental retardation or dementia following slow deterioration in intellectual abilities in the 20s occurs in over 50% of individuals [ Verrips, van Engelen, ter Laak et al 2000 ]. Some individuals show mental impairment from early infancy, whereas the majority have normal or only slightly subnormal intellectual function until puberty. In the spinal form, intellect is almost always normal.
Neuropsychiatric symptoms such as behavioral changes, hallucinations, agitation, aggression, depression, and suicide attempts may be prominent.
Pyramidal signs (i.e., spasticity) and/or cerebellar signs are almost invariably present between 20-30 years of age. A spinal form, in which spastic paraparesis is the main clinical symptom, was described by van Bogaert ( 1962 ) and more recently by Verrips, Nijeholt et al ( 1999 ).
Extrapyramidal manifestations including dystonia and atypical parkinsonism have been reported on occasion [ Fiorelli et al 1990 , Rogelet et al 1992 , Dotti et al 2000 , Grandas et al 2002 ]. Although palatal myoclonus was observed in the first individual reported [van Bogaert et al 1937 ], it was not observed in a large series of affected individuals [ Dotti et al 2001 ].
Seizures are reported in about 50% of individuals with CTX [ Matsumuro et al 1990 , Arlazoroff et al 1991 , Dotti et al 1996 ].
Peripheral neuropathy is evident on electrophysiological studies [ Ohnishi et al 1979 , Argov et al 1986 , Federico et al 1987 , Ben Hamida et al 1991 ], which reveal decreased nerve conduction velocities (NCV) and abnormalities in somatosensory, motor, brainstem, and visual evoked potentials [ Mondrelli et al 1992 ]. Clinical manifestations related to peripheral nerve involvement are distal muscle atrophy and pes cavus. Sensory abnormalities are rarely described.
Neuropathology. Classic CNS pathology findings in CTX include granulomatous and xanthomatous lesions in the cerebellar hemispheres, globus pallidus, and cerebellar peduncles. Demyelination and gliosis and involvement of the long tract of the spinal cord have been described [ van Bogaert et al 1937 , van Bogaert 1962 ]. Nerve biopsy reveals primary axonal degeneration, demyelination, and remyelination. Federico et al ( 1991 ) found mild myopathic changes of increased variability of fiber size with randomly distributed atrophic fibers. Ultrastructural abnormalities included mitochondrial subsarcolemmal aggregates and morphological changes of these organelles [ Federico et al 1991 ]. Reduced respiratory chain enzyme activity has been observed [ Dotti et al 1995 ].
Neuroimaging. Changes on CT and MRI include diffuse brain and cerebellar atrophy, white matter signal alterations, and bilateral focal cerebellar lesions [ Berginer et al 1981 , Waterreus et al 1987 , Berginer et al 1994 , Dotti et al 1994 , De Stefano et al 2001 ]. MR spectroscopy shows decreased n-acetylaspartate and increased lactate indicative of widespread brain mitochondrial dysfunction [ De Stefano et al 2001 ]. The quantitative assessment of brain damage in CTX with use of magnetization transfer MR imaging has been recently described [ Inglese et al 2003 ].
Genotype-Phenotype Correlations
Several authors have attempted to correlate genotype to phenotype , but no correlation has been identified [ Dotti et al 1996 , Verrips, van Engelen, Wevers et al 2000 ]. The interaction of many genes and other factors may influence the clinical presentation.
Prevalence
The prevalence is not known.
Series of affected individuals have been reported in Israel and the USA [ Berginer et al 1984 ], Italy [ De Stefano et al 2001 , Dotti et al 2001 ], Japan [ Kuriyama et al 1991 ], and the Netherlands [ Waterreus et al 1987 ; Verrips, Hoefsloot et al 2000 ]. Affectedindividuals have been reported in Belgium [ van Bogaert et al 1937 , Philippart et al 1969 ], Brazil [ Canelas et al 1983 ], Canada [ Pastershank et al 1974 ], France [ Rogelet et al 1992 ], Iran [ Farpour & Mahloudji 1975 ], Norway [ Schreiner et al 1975 ], Tunisia [Ben Hamida et al 1991 ], Spain [ Campdelacreu et al 2002 ], China [ Ko & Lee 2001 ], and Sweden [ Rystedet et al 2002 ].
Differential Diagnosis
For current information on availability of genetic testing for disorders included in this section, see GeneTests Laboratory Directory . —E D .
Differential diagnosis of xanthomas include:
- Sitosterolemia, inherited sterol storage disease characterized by tendon xanthomas and by a strong predisposition to premature atherosclerosis. Serum concentration of plant sterols (sitosterol and campesterol) is increased. Primary neurological signs and cataracts are not present. Spastic paraparesis may occur as a result of spinal cord compression by multiple intradural, extramedullary xanthomas [ Hatanaka et al 1990 ].
- Hypercholesterolemia and hyperlipemia (especially type IIa), in which plasma cholestanol level is normal
When xanthomas are not evident, the differential diagnosis includes all forms of progressive mental deterioration [ Gilad et al 1999 ; Verrips, van Engelen, ter Laak et al 2000 ].
Early-onset cataract. Cruysberg ( 2002 ) reports that CTX comprises the second largest group of individuals with early-onset cataract and known neurologic disease. ( Myotonic dystrophy is the largest group.) Unexplained juvenile-onset cataracts associated with infantile-onset chronic diarrhea and mental retardation or deterioration strongly suggest the possibility of CTX [ Cruysberg et al 1991 , Cruysberg et al 1995 ; Verrips, van Engelen, ter Laak et al 2000 ].
Management
Clinical improvement of individuals with CTX following chenodeoxycholic acid treatment (CDCA, a drug extensively used in the treatment of bile acid metabolism with cholesterol gallstones) was first reported by Berginer et al ( 1984 ). Long-term CDCA treatment (750mg/day in adults) normalizes bile acid synthesis, leading to disappearance of abnormal metabolites from serum, bile, and urine; normalizes plasma and CSF concentration of cholestanol by suppressing cholestanol biosynthesis; and improves neurophysiological findings [ Mondelli et al 1992 , Mondelli et al 2001 ] and other clinical manifestations including osteoporosis [ Federico et al 1993 ]. In a study of 11 years of treatment with CDCA, Mondelli et al ( 2001 ) reported that four months into treatment, nerve conduction velocities normalized and subsequently remained stable; motor evoked potentials (MEPs) and sensory evoked potentials (SEPs) improved slowly but continuously; and clinical manifestations stabilized, but neurologic deficits did not improve. The contrast between two untreated siblings whose symptoms progressed and a third treated sibling whose symptoms stabilized suggests that treatment is beneficial.
Although CDCA is considered the best treatment for CTX [ Samenuk & Koffman 2001 ], it has recently ceased to be available as other more effective drugs for gallstones have been utilized; its unavailability has left affected individuals without an essential drug for their disease, with the exception of affected individuals in Italy, who are closely monitored [ Federico & Dotti 2001 ].
Inhibitors of HMG-CoA reductase alone or in combination with CDCA are also effective in decreasing cholestanol concentration and improving clinical signs [ Peynet et al 1991 , Verrips et al 1999 ]. However, because of clinical evidence that HMG-CoA reductase inhibitors may induce muscle damage and even rhabdomyolysis, caution is required in the use of these drugs [ Federico & Dotti 1994 ].
Other possible treatments include low-density lipoprotein (LDL) apheresis, but the results are controversial [ Mimura et al 1993 , Berginer & Salen 1994 ].
Liver transplantation, although never performed in individuals with CTX, remains a possibility.
Eyes. Cataract extraction is typically required in at least one eye by age 50 years.
Symptomatic treatments for epilepsy, spasticity, and parkinsonism have been utilized. Parkinsonism is poorly responsive to levodopa, whereas an antihistamine drug, diphenylpyraline hydrochloride, had an excellent effect in three individuals [ Ohno et al 2001 ].
Genetic Counseling
Genetic counseling is the process of providing individuals and families with information on the nature, inheritance, and implications of genetic disorders to help them make informed medical and personal decisions. The following section deals with genetic risk assessment and the use of family history and genetic testing to clarify genetic status for family members. This section is not meant to address all personal, cultural, or ethical issues that individuals may face or to substitute for consultation with a genetics professional. —E D .
Mode of Inheritance
Cerebrotendinous xanthomatosis is inherited in an autosomal recessive manner.
Risk to Family Members
This section is written from the perspective that molecular genetic testing for this disorder is available on a research basis only and results should not be used for clinical purposes. This perspective may not apply to families using custom mutation analysis . —E D .
Parents of a proband
- The parents of an affected child are obligate heterozygotes , and, therefore, carry one mutant allele .
- Heterozygotes ( carriers ) are generally asymptomatic, although an increased incidence of cardiovascular disorders and gall stones has been observed in obligate carriers [personal observation].
Sibs of a proband
- At conception, the sibs of an affected individual have a 25% chance of being affected , a 50% chance of being asymptomatic carriers , and a 25% chance of being unaffected and not carriers .
- Once an at-risk sib is known to be unaffected , the chance of his/her being a carrier is 2/3.
- Heterozygotes ( carriers ) are generally asymptomatic.
Offspring of a proband . The offspring of an individual with CTX are obligate heterozygotes ( carriers ) for a disease-causing mutation in the CYP27A1 gene .
Other family members of a proband . Sibs of the proband’s parents are at 50% risk of being carriers .
Carrier Detection
Carrier testing using molecular genetic techniques is not offered because it is not clinically available.
Related Genetic Counseling Issues
Disease manifestations in heterozygotes . An increased frequency of cardiovascular disorders and gallstones has been observed in families of affected individuals [personal observation].
Testing of at-risk children. Early diagnosis of at-risk family members allows initiation of treatment that may prevent or limit disease manifestations.
Family planning. The optimal time for determination of genetic risk is before pregnancy.
DNA banking . DNA banking is the storage of DNA (typically extracted from white blood cells) for possible future use. Because it is likely that testing methodology and our understanding of genes , mutations , and diseases will improve in the future, consideration should be given to banking DNA of affected individuals. DNA banking is particularly relevant in situations in which molecular genetic testing is available on a research basis only. See DNA Banking for a list of laboratories offering this service.
Prenatal Testing
No laboratories offering molecular genetic testing for prenatal diagnosis of CTX are listed in the GeneTests Laboratory Directory. However, prenatal testing may be available for families in which the disease-causing mutations have been identified in an affectedfamily member in a research or clinical laboratory. For laboratories offering custom prenatal testing .
Author Information
Antonio Federico, MD
Neuroscience
Maria Teresa Dotti, MD
Neuroscience
University of Siena
Antonio Federico is a full professor of Clinical Neurology and Director of the Department of Neurological and Behavioral Sciences, University of Siena. Maria Teresa Dotti is an associate professor at the University of Siena.